Taking the fear out of FDA UDI requirements.
UDI Compliance Guide
Contributed by | Microscan Systems, Inc.
UDI Compliance Guide
Meeting compliance requirements for the FDA Unique Device Identification (UDI) rule is priority-one for today’s medical device manufacturers and labelers. With compliance dates on the horizon for new classes of devices, businesses must be proactive in ensuring standardized product serialization and update their labeling procedures at a rapid pace, or forfeit the right to participate legally in the marketplace. The pressure to meet this demand can be daunting, and understanding the available UDI documentation can also be a formidable challenge to the new UDI adopter. This whitepaper offers guidelines for simplifying each step toward UDI compliance and answers common questions about the UDI rule, such as:
- What is Unique Device Identification?
- Who is required to participate?
- What is the difference between a DI and a PI?
- How do I register with the FDA and GUDID?
- What is the timeline to become compliant?
- What are the different classes of devices and their requirements?
What is Unique Device Identification?
- The FDA has established a unique device identification system to identify and trace medical devices throughout their distribution and use. The UDI Final Rule was published 24 September 2013.
- As part of this mandate, labels on medical devices must contain a unique device identifier (UDI) code in human- and machine-readable form. The machine-readable form must be able to be interpreted by automatic identification and data capture (AIDC) technology; that is, a barcode or machine-readable text that can be decoded by a barcode reader or smart camera.
- Labelers must also submit certain information about each device to FDA’s Global Unique Device Identification Database (GUDID). The GUDID is maintained by the U.S. Food and Drug Administration, and only contains a portion of the UDI known as the DI (Device Identifier). Other portions of the UDI including PIs (Production Identifiers) and the data within production identifiers remain with the manufacturer. The GUDID is not intended to replace accurate recordkeeping done by device manufacturers, but act as a reference catalog for every device with an identifier and a de-duplication method for all UDIs.
- UDI is expected to improve greatly upon patient safety and healthcare industry processes by implementing a global system of standards which is fundamental to enabling an effective and efficient implementation of the UDI rule by all healthcare stakeholders worldwide.
The FDA UDI rule applies to labelers. Who is considered a labeler?
The requirements of the FDA UDI rule applies to labelers of medical devices. Currently, only these labelers are responsible for meeting requirements for UDI labeling and GUDID data submission.
A “labeler” is defined as:
A. Any person or agency who causes a label to be applied to a device with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label; and,
B. Any person or agency who causes the label of a device to be replaced or modified with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label. The addition of the name of, and contact information for, a person who distributes the device, without making any other changes to the label, is not a modification for the purposes of determining whether a person is a labeler. In most instances, the labeler is the device manufacturer, but the labeler may also be a specification developer, a single-use device re-processor, a convenience kit assembler, a re-packager, or a re-labeler.
The UDI rule primarily deals with device labelers' development of UDIs, their placement on device labels and packages, and submission of this information to the GUDID. Full UDI implementation will always require other stakeholders – including physicians, hospitals, payers, and patients – to integrate UDIs in their respective systems. Distributors and providers of inventory management systems should also have the ability to capture and transmit UDIs so that device-specific information can flow seamlessly from manufacturer to provider.
Does the UDI rule apply to medical device manufacturers outside of the U.S.?
The U.S. FDA rules apply to any medical devices sold in the U.S. International medical device manufacturers must comply with these rules as well if the device is being sold in the U.S., regardless of where the device is manufactured.
What is a UDI?
A UDI is a unique numeric or alphanumeric code, which consists of two parts:
1. Device Identifier (DI) – a mandatory, fixed portion that identifies the labeler and the specific version or model of a device. This portion is required and must be submitted to the GUDID database.
2. Production Identifier (PI) – a conditional, variable portion of a UDI. This information is maintained by the manufacturer for internal operations and is not submitted to the GUDID database.

A fictitious example of a medical device label that conforms to the requirements of the FDA’s UDI (Unique Device Identifier) initiative for device labels and packaging.
What is a Production Identifier (PI)?
The production identifier (PI) identifies one or more of the following pieces of data when included on the label of a device:
- Lot or batch number within which a specific device was manufactured.
- Serial number of a specific device.
- Expiration date of a specific device.
- Date a specific device was manufactured.
The FDA UDI rule does not require that specific PIs be included in the UDI of a particular device. However, it requires that whatever PIs appear on the device label or package also be included in the UDI in both human- and machine-readable format. For example, if a label includes the Expiration Date, Batch Number, and Serial Number of a device, all of this information must also be included in the UDI that is printed on the device’s label.
Where do I get a Device Identifier (DI)?
For the purpose of maintaining standardization across all devices bearing UDI, a manufacturer’s DI must be issued under a system operated by an FDA-accredited issuing agency such as:
- GS1: www.gs1.org
- Health Industry Business Communications Council (HIBCC): www.hibcc.org
- ICCBBA: www.iccbba.org
Once received, each DI must be provided on the label as part of the UDI in both human- and machine-readable format. Formatting requirements vary by the issuing agency and must be strictly adhered to in order to meet compliance for the agency’s specifications. It is important to understand the specifications of each issuing agency and establish a testing process to ensure UDIs continue to comply. Testing can be accomplished using technology like a barcode verifier or other standards-based verification method.

Once you have selected an accredited issuing agency a critical part of moving forward successfully is understanding the standards. All three barcodes above are Code 128 symbols; however, each issuing agency has their own specifications on how the data is to properly be encoded to comply.
How many DIs am I required to have?
A DI is comprised of a unique Company Identifier and a unique number assigned to a specific model or version of a manufactured device. If you are manufacturing and labeling devices and you have 20 different models or versions, you would need to obtain 20 unique Device Identifiers (DIs), one for each version.
I received my DI. What do I do now?
In order to ensure UDI compliance, labelers must submit their DI to the GUDID. To do this, labelers must first request a GUDID account. Beginning January 26, 2015, the FDA began to accept GUDID account requests from labelers of implantable, life-supporting, and life-sustaining (I/LS/LS) devices. The FDA will also continue to accept GUDID accounts from labelers of Class III devices and devices licensed under the Public Health Service Act (PHS Act). These labelers are encouraged to obtain a GUDID account as soon as possible to allow adequate time for system compatibility testing and data submission.
The GUDID account identifies the labeler in GUDID and is required for all labelers under the UDI rule.
Labelers must submit a GUDID account request on the FDA website: http://www.fda.gov.
To comply with federal regulations, it is also important to ensure that the proper procedures are in place to make compliance an intrinsic part of everyday operations. You may want to start with your company’s QMS (Quality Management System) to make sure that product serialization requirements are updated to include the new requirements of the UDI rule. Typically, when labeling or packaging for a product is changed, there is a change order process that must be adhered to, which follows what is outlined in the QMS. New or updated process and procedures guidelines should be implemented to control how UDI is administered (created and applied to labels) and how UDI numbers will be managed (kept on record for future reference). This may include processes related to labeling, recall activity, traceability records, supplier management, and customer complaints.
What kind of device packaging is required to bear a DI?
Per the UDI label and GUDID submission requirements, a “device package” is a package that contains a fixed quantity of a unique version or model of a device. In order to identify a device throughout distribution and use, the various packaging (material used to group each unique fixed quantity, from unit level to case level) must have a unique identifier. If a device is sold in a unit-level device package (bottle, box, or other unit-level package), and then packaged as a box of 20, and then ten boxes of 20 are shipped in the carton, each level of packaging would require its own Device Identifier.

Is my device required to have a UDI directly marked on the device itself?
The permanence of a UDI on a device depends on the type of device you are manufacturing. Medical devices that are intended to be used more than once or are intended to be re-processed prior to each use must have a directly-marked UDI (this is called a direct part mark, or DPM). Usually this is applied in the form of a barcode etched or abraded onto the surface of a device to ensure the device’s identifying information is not lost over time.
The FDA has stated that implantable devices will not require a direct part mark, nor will single-use devices that are disposed of after use.
I already have a Labeler Code. Can I still use it?
The FDA permits the continued use of an NDC/NHRIC Labeler Code issued under an FDA-accredited system as an identifier in the UDI. The use of the Labeler Code must be approved by the issuing agency that administrates the system (for instance, GS1 or HIBCC) and the labeler must submit a request for continued use of the Labeler Code. The FDA must have received the request no later than September 24, 2014.
The FDA UDI rule does sunset the use of NDC/NHRIC codes for medical devices on the date that a device is required to be labeled with the UDI. After this date, however, a labeler can no longer continue to label a device with the NHRIC number. Prior to the date of required UDI compliance for your device, a labeler is allowed to label a device with both a UDI and NHRIC. In the GUDID database, it is suggested that you enter the UDI as the primary DI with the NHRIC as the secondary DI, and then remove the secondary DI prior to your compliance date.
What is the timeline to become UDI compliant?
The UDI system will go into effect in stages over a period of seven years to ensure a smooth implementation and to spread the costs and burdens of implementation over time.
|
Compliance Date |
Device Class |
What kind of devices? |
UDI Application |
Requirement |
|
September 24, 2014 |
Class III Medical Devices; Devices under the Public Health Service Act (PHS Act) |
Greatest risk upon failure. Support or sustain human lives; malfunction is unacceptable. Pacemakers, heart valves, implants, etc. |
Printed on labels and packaging |
The labels and packages of Class III medical devices and devices licensed under the Public Health Service Act (PHS Act) must bear a UDI, which must be submitted to the GUDID database. |
|
Class III Stand-Alone Software |
Greatest risk upon failure. Support or sustain human lives; malfunction is unacceptable. Pacemakers, heart valves, implants, etc. |
Plain-text statement in software |
Class III stand-alone software must submit its UDI to the GUDID. Regardless of whether it is or is not distributed in packaged form, stand-alone software regulated as a medical device must provide its unique device identifier through either or both of the following: (1) An easily-readable, plain-text statement displayed whenever the software is started; (2) An easily-readable, plain-text statement displayed through a menu command (e.g., an “About * * *” command). |
|
|
September 24, 2015 |
Implantable, Life-Supporting, and Life-Sustaining Devices* |
Greatest risk upon failure. Support or sustain human lives; malfunction is unacceptable. Pacemakers, heart valves, implants, etc. |
Printed on labels and packaging; permanent marking on the device |
The labels and packages of implantable, life-supporting, and life-sustaining devices must bear a UDI, which must be submitted to the GUDID database.
A device that is a life-supporting or life-sustaining device that is required to be labeled with a UDI must a bear UDI as a permanent marking on the device itself (i.e., DPM code) if the device is intended to be used more than once and intended to be reprocessed before each use. This UDI data must be submitted to the GUDID database. |
|
Stand-Alone Software that is a Life-Supporting or Life-Sustaining Device* |
Greatest risk upon failure. Support or sustain human lives; malfunction is unacceptable. Pacemakers, heart valves, implants, etc. |
Plain-text statement in software |
Stand-alone software that is a life-supporting or life-sustaining device must submit its UDI to the GUDID. Regardless of whether it is or is not distributed in packaged form, stand-alone software regulated as a medical device must provide its unique device identifier through either or both of the following: (1) An easily-readable, plain-text statement displayed whenever the software is started; (2) An easily-readable, plain-text statement displayed through a menu command (e.g., an “About * * *” command). |
|
|
September 24, 2016 |
Class III Devices
|
Greatest risk upon failure. Support or sustain human lives; malfunction is unacceptable. Pacemakers, heart valves, implants, etc. |
Permanent marketing on the device |
Class III devices required to be labeled with a UDI must bear a UDI as a permanent marking on the device itself (i.e., DPM code) if the device is a device intended to be used more than once and intended to be reprocessed before each use. |
|
Class II Devices
|
Minimal risk upon failure. Complex design, minimal risk. X-ray machines, powered wheelchairs, infusion pumps, surgical, acupuncture needles. |
Printed on labels and packaging |
The labels and packages of Class II medical devices must bear a UDI, which must be submitted to the GUDID database. |
|
|
Class II Stand-Alone Software |
Minimal risk upon failure. Complex design, minimal risk. X-ray machines, powered wheelchairs, infusion pumps, surgical, acupuncture needles. |
Plain-text statement in software |
Class II stand-alone software must submit its UDI to the GUDID. Regardless of whether it is or is not distributed in packaged form, stand-alone software regulated as a medical device must provide its unique device identifier through either or both of the following: (1) An easily-readable, plain-text statement displayed whenever the software is started; (2) An easily-readable, plain-text statement displayed through a menu command (e.g., an “About * * *” command). |
|
|
September 24, 2018 |
Class II Devices
|
Minimal risk upon failure. Complex design, minimal risk. X-ray machines, powered wheelchairs, infusion pumps, surgical, acupuncture needles. |
Permanent marketing on the device |
A Class II device that is required to be labeled with a UDI must bear a UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. This UDI data must be submitted to the GUDID database. |
|
Class I Devices; |
Little to no risk upon failure. Tongue depressors, elastic bandages, handheld dental instruments, examination gloves. |
Printed on labels and packaging |
The labels and packages of Class I medical devices and devices that have not been classified into Class I, Class II, or Class III must bear a UDI, which must be submitted to the GUDID database. |
|
|
Class I Stand-Alone Software |
Little to no risk upon failure. Tongue depressors, elastic bandages, handheld dental instruments, examination gloves. |
Plain-text statement in software |
Class I stand-alone software must submit its UDI to the GUDID. Regardless of whether it is or is not distributed in packaged form, stand-alone software regulated as a medical device must provide its unique device identifier through either or both of the following: (1) An easily-readable, plain-text statement displayed whenever the software is started; (2) An easily-readable, plain-text statement displayed through a menu command (e.g., an “About * * *” command). |
|
|
September 24, 2020 |
Class I Devices; |
Little to no risk upon failure. Tongue depressors, elastic bandages, handheld dental instruments, examination gloves. |
Permanent marking on the device |
Class I devices, and devices that have not been classified into Class I, Class II, or Class III that are required to be labeled with a UDI, must a bear UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. This UDI data must be submitted to the GUDID database. |
* The FDA recommends that labelers search the CDRH Product Classification database for the most current information on FDA product codes to determine if devices are considered implantable, life-sustaining, or life-supporting.
Full compliance dates and requirements can be viewed at www.fda.gov.
What are Class I, Class II, and Class III devices?
The FDA classifies medical devices based on the risks associated with the device. Devices are classified into one of three categories—Class I, Class II, and Class III.
- Class I devices are deemed to be low risk and are therefore subject to the least regulatory controls. For example, dental floss and gauze bandages are classified as Class I devices.
- Class II devices are higher risk devices than Class I and require greater regulatory controls to provide reasonable assurance of the device’s safety and effectiveness. For example, condoms are classified as Class II devices.
- Class III devices are generally the highest risk devices and are therefore subject to the highest level of regulatory control. Class III devices must typically be approved by FDA before they are marketed. For example, replacement heart valves and other implantable devices are classified as Class III devices.
Suggestions for Smooth UDI Implementation
1. Select your issuing agency and read the agency’s specifications carefully.
2. Take note of any device revision, model variation, or packaging level in your manufacturing process that requires a UDI.
3. Request DIs from the issuing agency for each unique device or package.
4. Once you receive your approved UDI from the issuing agency, request a submission to the GUDID.
5. Select your data carrier. A data carrier is a machine-readable element such as a barcode or other symbol that encodes the UDI. Confirm that the data carrier is allowed by the issuing agency for your application.
6. When creating a barcode, select barcode software that has the capability of supporting the issuing agency requirements.
7. During the creation of a new label, use a barcode verifier to ensure that the data in your barcode is properly structured according to the issuing agency specifications. To do this, create a label, then print the label to a desktop printer. Test the structure and quality of barcodes on your printed label using a verifier that offers software parameters for the specific issuing agency specifications you are using.
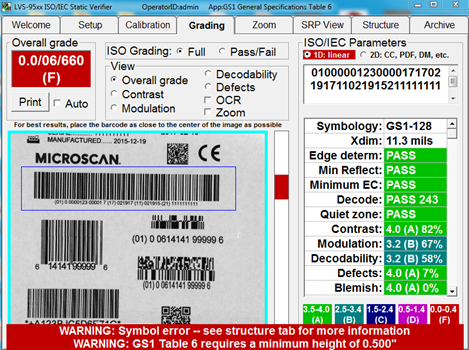
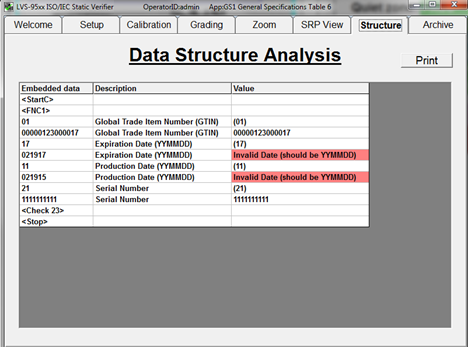
Catch barcode structure and readability errors by testing labels with standards-based verification software before sending to production. Some errors that may cause noncompliance are not always visible to the naked eye. This verification software offers built-in parameters that ensure GS1 barcodes are compliant with the specifications for each type of device.
8. Maintain the aspect ratio of your barcode after creation. Verify your label again after the barcode has been incorporated into label artwork and make sure that the barcode (data carrier) has not been compromised.

9. When you have validated that the barcode structure is compliant, begin the printing process. During printing, you may use an in-line verification system (integrated onto your line or into your printer) to ensure that print quality for your label is maintained to the standards required by your issuing agency (such as print quality standards like ISO/IEC 15415 or ISO/IEC 15416).

Conclusion
Although there are a number of steps toward ensuring UDI compliance, and a number of ways that these changes impact internal operations, there is a definable path to follow for implementation. The guidance offered by this paper is intended to clarify the information surrounding FDA UDI and simplify the path to compliance, but each application and issuing agency brings its own UDI considerations. It is important to understand your requirements when it comes to device classes, timelines for compliance, and identifiers required for your device and issuing agency. If needed, personalized guidance can be acquired from professional product serialization, FDA UDI compliance, and label verification agencies. These subject matter and technology experts can support the transition to UDI compliance for your medical devices and act as consultants for additional assistance with implementation.
References
- FDA. (2015, May 06). Compliance Dates for UDI Requirements. Retrieved February 12, 2016, from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/CompliancedatesforUDIRequirements/default.htm
- FDA. (2016, February 10). Request a GUDID Account. Retrieved February 12, 2016, from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/ucm416113.htm
- FDA. (2015, May 06). UDI Basics. Retrieved February 12, 2016, from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/UDIBasics/default.htm
- FDA. (2015, December 28). What does it mean for FDA to “classify” a medical device?. Retrieved February 12, 2016, from http://www.fda.gov/AboutFDA/Transparency/Basics/ucm194438.htm
- [Untitled image of example of a unique device identifier (UDI) on a medical label]. (2013, September 18). Retrieved February 12, 2016 from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/UDIBasics/default.htm
The content & opinions in this article are the author’s and do not necessarily represent the views of ManufacturingTomorrow
Comments (0)
This post does not have any comments. Be the first to leave a comment below.
Featured Product

